联系方式:400-996-8673(微信同号)
地址:北京市海淀区清华同方科技大厦
 扫描下方二维码 添加工程师微信
扫描下方二维码 添加工程师微信

不同催化剂的析氢性能研究——清析客户检测案例
类别:客户案例
 2180
2180
 2024-03-12
2024-03-12
2180
2024-03-12
2024年1月12日,本人接到兰州某大学的计算需求。接到反馈后,第一时间与客户进行详细沟通,具体情况如下:
客户背景
客户是兰州某大学的教授,希望通过模拟计算软件,计算Pt单质表面模型、Ru-O4表面模型、Ru团簇负载的Ru-O4表面模型、Ru团簇负载的Ru-N4表面模型对于析氢反应的吉布斯自由能计算。
样品名称
Pt单质表面模型

Ru-O4表面模型
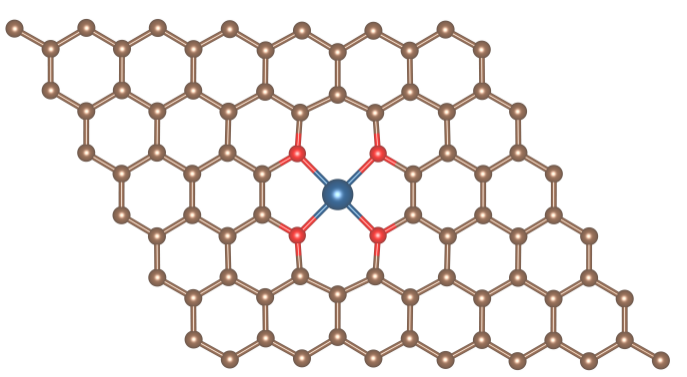
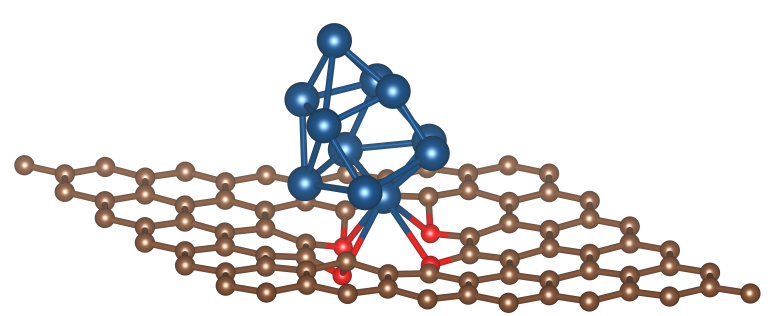
Pt团簇负载的Ru-O4表面模型
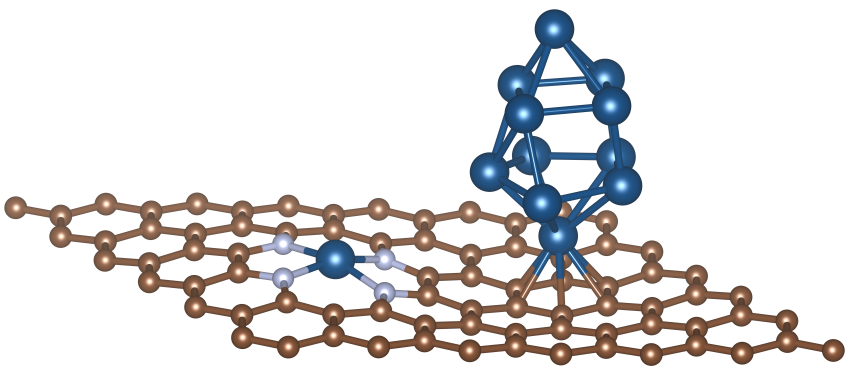
Ru团簇负载的Ru-N4表面模型
客户需求
1. 客户希望通过计算模型3计算 Ru团与基底Ru 原子之间的电荷密度差分研究具体的电子转移情况。
2. 模型 1、模型2、模型3计算 DOS (特别: 提取模型 3的Ru 团和基底R 原子的 d带中心数据) 希望通过计算CN,CN-Co和CoN4P1构型材料DOS对比,说明带隙缩短,P原子掺杂导致中间带隙产生,Co原子贡献在CB, CBM处态密度上升,表明更多电子可以被激发到CB。
3. 模型3、模型4、模型5计算碱性 HER 反应路径,如果下图所示 实验组模型(一步双电子,涉及1O2):Co-N3P1材料和氧气分离→氧气共振激发成1O2→单线态氧快速提取两个电子和两个H+形成过氧化氢。
对照组模型(两步单电子,涉及O2·-):Co-N4材料和氧气分离→氧气吸附在材料表面→氧气得到一个电子和氢质子形成*OOH(超氧自由基)→氧气再得到一个电子和氢质子形成过氧化氢。
解决方案
所有的DFT计算均基于维也纳从头计算模拟软件包(VASP)[1-2]进行。交换相关势由Perdew-Burke-恩泽霍夫(PBE)广义梯度方法(GGA)[3]描述。电子-离子的相互作用是由投影仪增广波(PAW)[4]来解释。所有的DFT计算都在450 eV的截止能量下进行,布里渊区使用3×3×1k点网格进行采样。自洽迭代的能量收敛准则和力的收敛准则分别设置为10-5 eV和0.02 eV A-1。采用DFT-D3方法描述范德华(vdW)相互作用[5]。 该反应的吉布斯自由能变化(ΔG)的计算公式如下:
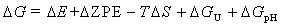
式中,ΔE为DFT计算直接得到的电子能差,ΔZPE为零点能差,T为室温(298.15 K),ΔS为熵变。ΔGU =-eU,其中U为外加电极电位。ΔGpH = kBT×ln 10×pH,其中kB为玻尔兹曼常数,pH值设为0。
(为保护客户隐私,测试结果不予展示)
客户反馈
数据结果符合预期,计算周期也与客户承诺一致。最后计算结果符合客户预期,客户对于计算结果非常满意,对工程师的专业度也很认可。

 9737
9737